Содержание
Турнир «Все свои»
13 этап турнира «ВСЕ СВОИ»
Регламент
Завершился 12 этап турнира «ВСЕ СВОИ»
Группа Challenger:
1 место — Богданов Валентин
2 место — Воробьёв Олег
3 место — Сидоренко Владимир
Группа Satellite:
1 место — Боднар Артём
2 место — Швед Станислав
3 место — Рудакова Наталья
Женская группа:
1 место — Топчий Светлана
2 место — Тихонова Ирина
3 место — Александрова Галина
Поздравляем победителей и благодарим всех участников турнира!
16 января 2022г завершился 11 этап турнира «ВСЕ СВОИ»
Группа Challenger:
1 место — Воробьёв Олег
2 место — Чувашов Константин
3 место — Сидоренко Владимир
Группа Satellite:
1 место — Богданов Валентин
2 место — Боднар Артём
3 место — Коломин Анатолий
Поздравляем победителей и благодарим всех участников турнира!
1 июня 2021г завершился десятый этап турнира «ВСЕ СВОИ»
Группа Masters:
1 место — Удальцов Михаил, 2 место — Кошевой Дмитрий, 3 место — Сапегин Егор.
Группа Challenger общая:
1 место — Воробьёв Олег, 2 место — Чусовитин Александр, 3 место — Сидоренко Владимир.
Группа Satellite:
1 место — Игнатов Максим, 2 место — Анненков Сергей, 3 место — Богданов Валентин.
Поздравляем победителей!
31 декабря 2020г завершился девятый этап турнира «ВСЕ СВОИ»
Группа Masters:
1 место — Кошевой Дмитрий, 2 место — Кочегаров Илья, 3 место — Тюрин Артём.
Группа Challenger общая:
1 место — Воробьев Олег, 2 место — Сапегин Егор, 3 место — Гражданова Ольга.
Группа Satellite:
1 место — Воробьев Денис, 2 место — Панов Константин, 3 место — Анненков Сергей.
Поздравляем победителей!
27 декабря завершился восьмой этап турнира «ВСЕ СВОИ»
Группа Masters:
1 место — Кошевой Дмитрий, 2 место — Удальцов Михаил, 3 место — Татьянкин Павел.
Группа Challenger общая:
1 место — Подымов Геннадий, 2 место — Шишкин Андрей, 3 место — Воробьев Олег.
Группа Satellite:
1 место — Тацкин Сергей, 2 место — Игнатов Максим, 3 место — Анненков Сергей.
Группа Challenger женская:
1 место — Субботина Ирина, 2 место — Александрова Галина, 3 место — Серафимович Ольга.
Поздравляем победителей!
26 мая 2019 завершился седьмой этап турнира «ВСЕ СВОИ»
Группа №1:
1 место — Соколов Сергей, 2 место — Ядрышников Артём, 3 место — Павленко Андрей
Группа №2:
1 место — Кошевой Дмитрий, 2 место — Истошин Александр, 3 место — Чусовитин Александр
Женская группа:
1 место — Александрова Галина, 2 место — Огурцова Татьяна, 3 место — Авдеева Татьяна
Поздравляем победителей!
15 декабря 2018г. завершился шестой этап турнира «Все свои»
завершился шестой этап турнира «Все свои»
Регламент клубного турнира «ВСЕ СВОИ». 6 этап
1. Вступительный взнос 1000 р. с Участника.(возвращается сертификатом* на 1000 р. на аренду тен.корта).
2. Система начисления очков: победитель – 3 очка, проигравший – 1 очко, техническое поражение (отказ от игры) — 0 очков. Если участник сыграл менее половины игр, то результаты игр с ним не учитываются при учете очков. В случае равенства очков у двух или нескольких участников по окончанию турнира места распределяются, исходя из лучшей разницы выигранных – проигранных геймов, в случае равенства этого показателя более высокое место занимает игрок с большим количеством выигранных геймов, если и здесь равенство, то проводится жеребьевка. При данных расчетах 3-ий сет (тай – брейк до 10 очков) приравнивается к гейму.
3.Турнир проводятся в 2 категориях:
3.1 Категория «Общая»
3. 2 Категория «Женщины»
2 Категория «Женщины»
В категорию «Общая» допускаются мужчины и женщины не зависимо от возраста. Для игр в этой категории участники делятся на две группы, при этом группы должны быть по возможности равного уровня игры с равным количеством игроков. Возможно участие одного участника в 2-х хгруппах. Максимальное количество игроков в группе – 11. . Игры проходят по круговой системе в каждой из групп. Затем проходят стыковые матч между группами по принципу – игроки , занявшие первые места в своих группах играют за «первое-второе» место в итоговом протоколе, вторые места – за «третье-четвёртое» место и т. д.В категории «Женщины» игры проходят по круговой системе с определением мест в итоговом протоколе. Максимальное количество участников в категории — 12, минимальное – 5.
3.3 Для распределения игроков по группам будет проведена жеребьевка
4. Встречи можно играть в любое свободное время (только не во время аренды участников по абонементу). Цена за 1 час составляет всегда 900 р.
Цена за 1 час составляет всегда 900 р.
5. Формат: игры из 3-х сетов, сеты до 6 геймов, 3 сет – тай — брейк до 10 очков. При счете «ровно» очко разыгрывается на «больше — меньше». При обоюдной договоренности соперников это правило может быть изменено на розыгрыш «решающего очка» Судейство самостоятельное.
6. Начало турнира 15.09.18г. Окончание турнира 15.12.18г.
Призы по итогам Турнира :
- От теннисного клуба « Луна» — кубки, дипломы, а также всем участникам — сертификаты на аренду тен.корта* (последнее место 1 час, предпоследнее 2 часа и т.д.)
- От теннисного магазина — ценные призы и подарки призёрам и участникам.
*Сертификаты не могут быть использованы для оплаты абонемента и проведения игры в рамках данного турнира. Срок действия сертификатов до 15.04.2019г.
Группа №1
Группа №2
Женская группа
Стыковые игры
Правила приёма в 2022-2023 учебном году
Документы — Правила приёма в 2022-2023 учебном году
Версия для слабовидящих
Документы
Файлы:
| СПИСОК АБИТУРИЕНТОВ |
|
|
| Создан Размер Скачиваний | 2022 20.  89 KB 89 KB207 | |||
| Приложение № 4 |
|
|
| Создан Размер Скачиваний | 2022 49 KB 73 | |||
| Правила приема в КОГПОАУ НПТ на 2022-2023 уч.год |
|
|
| Создан Размер Скачиваний | 2022 566.48 KB 160 | |||
| Административный регламент по предоставлению услуги Зачисление в профессиональную образовательную организацию |
|
|
| Создан Размер Скачиваний | 2022 685.  72 KB 72 KB54 | |||
| Административный регламент по предоставлению услуги о зачислении в образовательную организацию |
|
|
| Создан Размер Скачиваний | 2022 624.21 KB 55 | |||
| Приложение № 1 к Правилам приема (заявление) |
|
|
| Создан Размер Скачиваний | 2022 62 KB 70 | |||
| Приложение № 2 к Правилам приема (документы) |
|
|
| Создан Размер Скачиваний | 2022 36 KB 69 | |||
| Приложение № 3 к Правилам приема (перечень специальностей и профессий) |
|
|
| Создан Размер Скачиваний | 2022 39 KB 77 | |||
| Приложение № 5 к Правилам приема (общежития) |
|
|
| Создан Размер Скачиваний | 2022 37.  5 KB 5 KB62 | |||
| Приложение № 6 к Правилам приема (расписка) |
|
|
| Создан Размер Скачиваний | 2022 31.5 KB 59 | |||
| Приложение №7 к Правилам приема (образец договора) |
|
|
| Создан Размер Скачиваний | 2021 35.01 KB 168 | |||
| Заявление на поступление в техникум |
|
|
| Создан Размер Скачиваний | 2021 61.  5 KB 5 KB219 | |||
| Согласие на обработку персональных данных |
|
|
| Создан Размер Скачиваний | 2020 28.05 KB 322 | |||
| Информация о прохождении поступающими медосмотра |
|
|
| Создан Размер Скачиваний | 2018 317.59 KB 783 | |||
| Инструкция для абитуриентов по подаче электронного заявления |
|
|
| Создан Размер Скачиваний | 2018 1.  13 MB 13 MB904 | |||
Закон о праве на судебное разбирательство
- Список журналов
- Рукописи авторов HHS
- PMC7416898
Clin Cancer Res. Авторская рукопись; доступно в PMC 2020 10 августа.
Опубликовано в окончательной редакции как:
Clin Cancer Res. 2020 15 января; 26(2): 340–343.
Опубликовано онлайн 2019 г. 30 октября. DOI: 10.1158/1078-0432.ccr-19-2015
PMCID: PMC7416898
NIHMSID: NIHMS1541631
PMID: 31666248
Авторский Исчерпавшие стандартные методы лечения часто ищут доступ к экспериментальным препаратам. Однако часто такой доступ недоступен либо из-за отсутствия исследования, либо из-за отсутствия открытого места для набора в исследование, даже если само исследование открыто, либо из-за неспособности пациента соответствовать одному или нескольким критериям включения в исследование. . В таких условиях пациенты часто ищут доступ к исследуемым агентам вне исследования. Федеральный закон «Право на попытку» был принят для создания дополнительного пути, отличного от программы расширенного доступа или «сострадательного использования» FDA, с помощью которой пациенты могли получить доступ к исследуемым препаратам. Спустя год после того, как этот закон был подписан, сохраняется как ограниченная осведомленность о нем, так и значительная степень непонимания со стороны тех, кто о нем знает. Закон создает возможность для значительного облегчения введения вне исследования, когда пациент, врач и производитель согласны в отношении использования подходящего исследуемого агента вне исследования. Закон, однако, не дает пациенту права предъявлять требования ни к поставщику, ни к производителю лекарств, а также не требует от какой-либо организации финансового покрытия лекарств. Приемлемыми препаратами являются те, которые не одобрены FDA ни по какому показанию, завершили исследование фазы I, проходят базовое исследование и имеют действующий план регистрации.
. В таких условиях пациенты часто ищут доступ к исследуемым агентам вне исследования. Федеральный закон «Право на попытку» был принят для создания дополнительного пути, отличного от программы расширенного доступа или «сострадательного использования» FDA, с помощью которой пациенты могли получить доступ к исследуемым препаратам. Спустя год после того, как этот закон был подписан, сохраняется как ограниченная осведомленность о нем, так и значительная степень непонимания со стороны тех, кто о нем знает. Закон создает возможность для значительного облегчения введения вне исследования, когда пациент, врач и производитель согласны в отношении использования подходящего исследуемого агента вне исследования. Закон, однако, не дает пациенту права предъявлять требования ни к поставщику, ни к производителю лекарств, а также не требует от какой-либо организации финансового покрытия лекарств. Приемлемыми препаратами являются те, которые не одобрены FDA ни по какому показанию, завершили исследование фазы I, проходят базовое исследование и имеют действующий план регистрации. Мы рассматриваем конкретный закон с комментариями о его последствиях для улучшения доступа к исследуемым препаратам вне клинических испытаний.
Мы рассматриваем конкретный закон с комментариями о его последствиях для улучшения доступа к исследуемым препаратам вне клинических испытаний.
30 мая 2018 года президент Трамп подписал закон, широко известный как Закон о праве на судебное разбирательство (RTT). По прошествии первого года действия этого закона мнения в отношении его поддержки сильно разнятся. Однако слишком немногие в онкологическом сообществе и в других местах точно знают, что есть в законе, а что нет, что ясно и что неясно, и, в конечном счете, что означает закон, а что нет. В последующем обзоре мы рассматриваем эти вопросы, поскольку они касаются прав и обязанностей тех, кто оказывает онкологическую помощь, и тех, кто ее получает. Поскольку у большинства участников онкологического сообщества практически нет формального юридического образования, мы разбили наш анализ, чтобы объяснить структуру и текст закона в терминологии и контексте, которые, по нашему мнению, будут понятны поставщикам медицинских услуг, нашим пациентам и лицам, осуществляющим уход за ними. .
.
RTT состоит из трех разделов. Раздел 1 — название, Раздел 2 — сам закон, а Раздел 3 — комментарий под названием «Смысл Сената». Мы обсудим эти разделы последовательно, с комментариями о том, что ясно, а что, по нашему мнению, остается открытым для интерпретации. Формулировки, приведенные в кавычках в тексте ниже, дословно взяты из Закона. 1
Раздел 1:
Официальное название закона: «Закон Трикетта Вендлера, Фрэнка Монгиелло, Джордана Маклинна и Мэтью Беллины о праве на судебное разбирательство от 2017 года».
Раздел 2:
Это действующий закон, и он начинается с заявления о том, что этот закон, в целом, является поправкой к главе V Федерального закона о пищевых продуктах, лекарствах и косметических средствах и охватывается нынешним разделом 561B этого Закона, озаглавленного «Исследуемые препараты для использования подходящими пациентами». Федеральный закон о пищевых продуктах, лекарствах и косметических средствах (или глава 9 раздела 21 Кодекса Соединенных Штатов) был принят Конгрессом в 1938 году и подписан президентом Франклином Д. Рузвельтом. Он установил юридические полномочия FDA по надзору за безопасностью и соблюдением правил в отношении пищевых продуктов, лекарств и косметики, при этом в главе V особое внимание уделялось лекарствам и устройствам, а в части E этой главы подробно описывались общие положения и процесс утверждения, связанные с новыми лекарствами и устройствами. В рамках главы V, части E Федерального закона о пищевых продуктах, лекарствах и косметических средствах, раздел 561B (или раздел 21 Кодекса США 360bbb-0) является федеральным законом о праве на судебное разбирательство (RTT). 2
Рузвельтом. Он установил юридические полномочия FDA по надзору за безопасностью и соблюдением правил в отношении пищевых продуктов, лекарств и косметики, при этом в главе V особое внимание уделялось лекарствам и устройствам, а в части E этой главы подробно описывались общие положения и процесс утверждения, связанные с новыми лекарствами и устройствами. В рамках главы V, части E Федерального закона о пищевых продуктах, лекарствах и косметических средствах, раздел 561B (или раздел 21 Кодекса США 360bbb-0) является федеральным законом о праве на судебное разбирательство (RTT). 2
Первая строка, предшествующая разделу 1, представляет закон словами «Закон, разрешающий использование неутвержденных медицинских изделий пациентами, у которых диагностировано неизлечимое заболевание…». Это указывает, и последующий текст ясно дает понять, что этот Закон не применяется к лекарственным средствам, которые уже находятся на рынке по любому показанию, поскольку они больше не будут «исследуемыми» или «неутвержденными», и что пациенты с потенциально излечимым заболеванием не будут подходящими пациентами для рассмотрения вопроса о лечении в соответствии с этим Законом (см. ).
).
Таблица 1.
Краткое изложение Закона о праве на судебное разбирательство
| Требования к пациенту a. Опасное для жизни состояние b. Варианты стандартной обработки исчерпаны c. Невозможно участвовать в продолжающемся испытании d. Дать информированное согласие Требования к препарату a. Испытание фазы I завершено 90 057 b. Текущее основное исследование фазы II или III c. План активного развития для получения одобрения FDA d. Не одобрено ни для каких показаний Требование к врачу a. Иметь хорошую репутацию в лицензирующей организации или совете b. Подтвердить, что пациент не может участвовать в клиническом испытании рассматриваемого препарата c. Примите письменное информированное согласие пациента или уполномоченного представителя. д. Не получать компенсацию от спонсора/производителя Требования к спонсору/производителю a.  Соблюдать стандартные процедуры маркировки исследуемых препаратов, продвижения на рынок и возмещения прямых затрат. Соблюдать стандартные процедуры маркировки исследуемых препаратов, продвижения на рынок и возмещения прямых затрат. б. Предоставляйте в FDA ежегодный отчет об использовании препарата, включая количество поставленных доз, количество пролеченных пациентов, способы применения препарата и любые известные серьезные нежелательные явления. Обязательства и полномочия a. Отсутствие ответственности за решение производителя не предоставлять препарат b. Отсутствие ответственности за решение врача не назначать препарат c. Нет полномочий для какой-либо организации предоставлять покрытие лекарств или связанного с ними ухода d. Никакое положительное право не установлено для любого лица |
Открыть в отдельном окне
Раздел 2, подраздел (a)(1) содержит определения правомочного пациента. Чтобы пациент имел право на участие, у него должно быть диагностировано «угрожающее жизни заболевание или состояние», он должен «исчерпать утвержденные варианты лечения», быть «неспособным участвовать в клиническом испытании с участием подходящего исследуемого препарата» и должен предоставить лечащему врачу «письменное информированное согласие в отношении приемлемого исследуемого препарата».
Критерий 1:
Для определения «опасного для жизни заболевания или состояния» делается ссылка на Раздел 312.81 Раздела 21 Свода федеральных правил.
Критерий 2:
То, что подразумевается под словом «исчерпаны» в отношении утвержденных вариантов лечения, может быть интерпретировано по-разному. Мы интерпретируем это как означающее, что подходящий пациент должен был пройти все утвержденные виды лечения, которые можно было бы безопасно применять. Таким образом, пациент, выбравший исследуемый агент в соответствии с этим законом, не сможет сделать это до получения всех доступных одобренных стандартных методов лечения.
Критерий 3:
Следующей областью субъективности является определение «неспособности» участвовать в клиническом испытании препарата. Мы предполагаем, что «неспособный» может быть охарактеризован либо как несоответствие критериям приемлемости для участия в ином доступном клиническом испытании (другой тип опухоли, неадекватное функциональное состояние или функция органа и т. д.), либо как географически неспособный участвовать в испытании из-за удаленности. из дома. Еще одно предостережение заключается в том, что пациент не будет соответствовать критериям, если он или она откажется от участия в доступном испытании фазы III, в котором желаемое исследуемое лекарство не дается во всех группах, а вместо этого попытается применить этот закон для улучшения своего или ее шансы на получение исследуемого препарата.
д.), либо как географически неспособный участвовать в испытании из-за удаленности. из дома. Еще одно предостережение заключается в том, что пациент не будет соответствовать критериям, если он или она откажется от участия в доступном испытании фазы III, в котором желаемое исследуемое лекарство не дается во всех группах, а вместо этого попытается применить этот закон для улучшения своего или ее шансы на получение исследуемого препарата.
В подразделе (a)(1) также приводится информация о лечащем враче. Врач должен иметь хорошую репутацию в ответственной лицензирующей организации или совете и должен подтвердить, что пациент «не может» участвовать в клиническом исследовании с исследуемым агентом. Кроме того, в законе прямо указано, что врач не должен получать компенсацию напрямую от производителя за назначение исследуемого препарата.
Критерий 4:
Подходящий пациент должен «предоставить лечащему врачу письменное информированное согласие в отношении подходящего исследуемого препарата». Это поднимает ряд важных и пока еще нерешенных вопросов. Каковы стандарты для этого информированного согласия? Кто несет ответственность за обеспечение ее полноты и правильности? По определению, исследуемый препарат, полученный в соответствии с этим законодательством, не является частью исследования и, следовательно, не находится под ответственностью Институционального наблюдательного совета. Предположительно, лечащий врач, на которого в соответствии с этим законодательством возложена ответственность за получение письменного информированного согласия, будет нести ответственность за полноту и точность такого информированного согласия. По логике вещей, производитель должен предоставить пациенту и/или лечащему врачу либо форму согласия из исследования фазы I (см. ниже), либо материалы, необходимые для составления формы согласия, такие как брошюра исследователя. Такие материалы вместе с инструкцией по аптеке, вероятно, потребуются аптеке учреждения для обработки и подготовки исследуемого препарата к приему.
Это поднимает ряд важных и пока еще нерешенных вопросов. Каковы стандарты для этого информированного согласия? Кто несет ответственность за обеспечение ее полноты и правильности? По определению, исследуемый препарат, полученный в соответствии с этим законодательством, не является частью исследования и, следовательно, не находится под ответственностью Институционального наблюдательного совета. Предположительно, лечащий врач, на которого в соответствии с этим законодательством возложена ответственность за получение письменного информированного согласия, будет нести ответственность за полноту и точность такого информированного согласия. По логике вещей, производитель должен предоставить пациенту и/или лечащему врачу либо форму согласия из исследования фазы I (см. ниже), либо материалы, необходимые для составления формы согласия, такие как брошюра исследователя. Такие материалы вместе с инструкцией по аптеке, вероятно, потребуются аптеке учреждения для обработки и подготовки исследуемого препарата к приему. Если учреждение уже проводит или провело судебное разбирательство с агентом по расследованию, то эти материалы, скорее всего, уже находятся в распоряжении учреждения. Если же учреждение ранее не работало с препаратом, то потребуются материалы, которые часто тщательно охраняются соглашениями и контрактами о конфиденциальности. Предположительно, компания потребует этих мер защиты своей интеллектуальной собственности, прежде чем предоставить агента врачу или учреждению.
Если учреждение уже проводит или провело судебное разбирательство с агентом по расследованию, то эти материалы, скорее всего, уже находятся в распоряжении учреждения. Если же учреждение ранее не работало с препаратом, то потребуются материалы, которые часто тщательно охраняются соглашениями и контрактами о конфиденциальности. Предположительно, компания потребует этих мер защиты своей интеллектуальной собственности, прежде чем предоставить агента врачу или учреждению.
Далее в Разделе 2 законодательства в подразделе (a)(2) определяются критерии, которым должно соответствовать лекарство, чтобы оно соответствовало требованиям настоящего Закона.
Во-первых, препарат должен быть исследуемым препаратом (согласно существующему законодательству), для которого завершена фаза I испытаний. Хотя в Законе нет определения термина «завершено», мы бы интерпретировали его как означающее, что критически важная информация, ради которой было предпринято исследование фазы I, доступна для принятия решений пациентом и врачом. В связи с этим мы утверждаем, что разумное определение «завершенного» исследования фазы I будет включать следующее: исследование навсегда закрыто для включения новых пациентов, база данных заблокирована, максимально переносимая доза (МПД), рекомендуемая доза фазы II, были выявлены дозолимитирующие и не дозолимитирующие токсические эффекты, и эти данные были рассмотрены и проверены спонсором и представлены в FDA. Без этого нельзя было бы достоверно определить правильную дозу для лечения, а также потенциальные риски и преимущества, и нельзя было бы получить надлежащее информированное согласие.
В связи с этим мы утверждаем, что разумное определение «завершенного» исследования фазы I будет включать следующее: исследование навсегда закрыто для включения новых пациентов, база данных заблокирована, максимально переносимая доза (МПД), рекомендуемая доза фазы II, были выявлены дозолимитирующие и не дозолимитирующие токсические эффекты, и эти данные были рассмотрены и проверены спонсором и представлены в FDA. Без этого нельзя было бы достоверно определить правильную дозу для лечения, а также потенциальные риски и преимущества, и нельзя было бы получить надлежащее информированное согласие.
Во-вторых, в подразделе (а)(2) оговаривается, что подходящим препаратом является тот, «который не был одобрен или не лицензирован для какого-либо использования». Таким образом, лекарство, которое одобрено (мы интерпретируем это как одобренное FDA) для любого показания , не соответствует требованиям настоящего Закона. Например, ингибитор иммунных контрольных точек, одобренный только для лечения меланомы, не может применяться в соответствии с этим Законом для пациентов с раком поджелудочной железы.
Кроме того, в этом подразделе говорится, что для того, чтобы лекарство соответствовало критериям, оно должно «проходить клиническое исследование, которое должно стать основным основанием для заявления об эффективности в поддержку одобрения или лицензирования». Такое испытание обязательно будет испытанием фазы II или фазы III. В нем также говорится, что лекарство должно быть предметом активной заявки на исследование нового лекарственного средства (IND), и «активная разработка или производство которого продолжается и не было прекращено производителем или приостановлено». Эти критерии приемлемости лекарств приведены в .
Далее в Законе идет подраздел (b) Раздела 2, в котором излагаются конкретные кодексы и правила, которые не применяются к соответствующим лекарствам, если они поставляются в соответствии с RTT. Это включает освобождение от части 312 раздела 21 Свода федеральных правил, 3 , который содержит требования, касающиеся пути расширенного доступа FDA к исследуемым препаратам. Однако в подразделе (b) указывается, что производители, которые спонсируют лекарства в рамках RTT, должны по-прежнему соблюдать стандартные требования к исследуемым новым лекарствам в отношении маркировки, продвижения и возмещения прямых затрат, указанных в разделах 312.6, 312.7 и 312.8 (d) ( 1) раздела 21 Свода федеральных правил.
Однако в подразделе (b) указывается, что производители, которые спонсируют лекарства в рамках RTT, должны по-прежнему соблюдать стандартные требования к исследуемым новым лекарствам в отношении маркировки, продвижения и возмещения прямых затрат, указанных в разделах 312.6, 312.7 и 312.8 (d) ( 1) раздела 21 Свода федеральных правил.
Далее следует подраздел (c), озаглавленный «Использование клинических результатов». В нем говорится, что «Секретарь не может использовать клинический исход, связанный с использованием подходящего исследуемого препарата в соответствии с настоящим разделом, для задержки или неблагоприятного воздействия на рассмотрение или утверждение такого препарата… если только — Секретарь не принимает решение … что использование такого клинический исход имеет решающее значение для определения безопасности подходящего исследуемого препарата; или спонсор запрашивает использование таких результатов». Это обеспечивает некоторую, но не полную защиту компании, которая может быть обеспокоена тем, что нежелательное явление, связанное с препаратом, может подорвать процесс утверждения.
В подразделе (d) указано, что спонсор должен ежегодно представлять отчет об использовании препарата в соответствии с этим законом, включая: «количество поставленных доз, количество пролеченных пациентов, использование, для которого препарат был предоставлен, и любые известные серьезные нежелательные явления». В этом подразделе также говорится, что Секретарь обязан публиковать годовой отчет на веб-сайте FDA. Таким образом, несмотря на то, что явное намерение состоит в том, чтобы нежелательные явления в соответствии с настоящим Законом не влияли на процесс утверждения, у спонсора могут быть опасения относительно риска того, что нежелательные явления и степень их обнародования могут повлиять на процесс утверждения. Такие опасения могут повлиять на готовность спонсора предоставить лекарство в соответствии с настоящим Законом.
Следующий подраздел Раздела 2 озаглавлен «Отказ от ответственности». Первый пункт этого подраздела гласит, что «отсутствие ответственности…. предъявляет претензии производителю или спонсору, лицу, выписывающему рецепты или отпускающему лекарству, или другому физическому лицу» в отношении исследуемого лекарственного средства, применяемого в соответствии с настоящим Законом, за исключением случаев «безрассудного или преднамеренного неправомерного поведения, грубой небрежности или умышленного правонарушения в соответствии с любым применимым законодательством штата». Второй пункт гласит, что «Спонсор, производитель, лицо, назначающее рецепты, отпускающее лекарство или другое физическое лицо не несет ответственности за свое решение не предоставлять доступ к подходящему исследуемому лекарственному средству». Таким образом, RTT не обязывает врача, производителя или другую организацию предоставлять лекарство в соответствии с настоящим Законом. Таким образом, он не дает пациенту фактических право попробовать лекарство, а скорее облегчает предоставление подходящего лекарства подходящему пациенту, если и врач, и спонсор согласны сделать это.
Второй пункт гласит, что «Спонсор, производитель, лицо, назначающее рецепты, отпускающее лекарство или другое физическое лицо не несет ответственности за свое решение не предоставлять доступ к подходящему исследуемому лекарственному средству». Таким образом, RTT не обязывает врача, производителя или другую организацию предоставлять лекарство в соответствии с настоящим Законом. Таким образом, он не дает пациенту фактических право попробовать лекарство, а скорее облегчает предоставление подходящего лекарства подходящему пациенту, если и врач, и спонсор согласны сделать это.
Раздел 3:
В этом разделе, озаглавленном «Смысл Сената», содержится контекст для толкования закона. Он не включен напрямую в поправку 561b Федерального закона о пищевых продуктах, лекарствах и косметических средствах, в отличие от Раздела 2. Из семи включенных утверждений мы выделяем три, которые мы считаем наиболее важными.
Во-первых, «Сенат считает, что… [данный Закон] не устанавливает новое право или не изменяет существующее право или иным образом не устанавливает положительное право для какой-либо стороны или лица».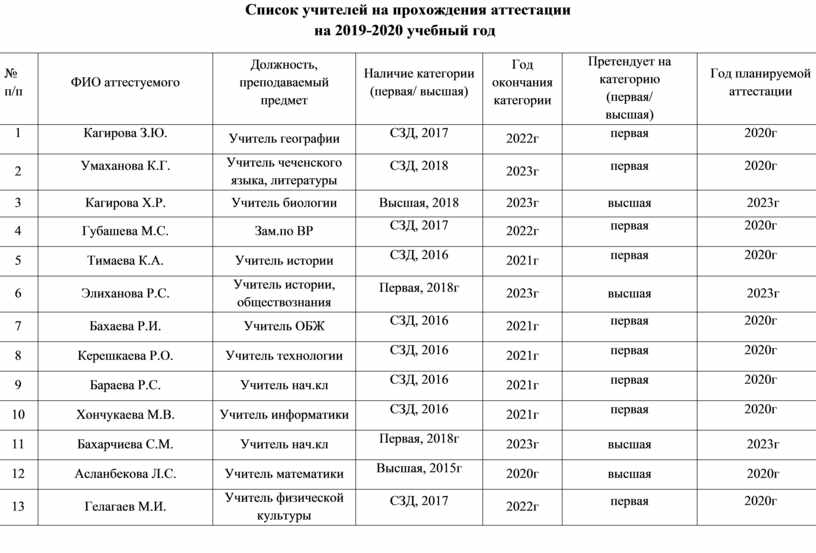 Следует отметить, что в соответствии с настоящим Законом не существует конкретных указаний относительно того, кто покрывает стоимость препарата или расходы на лечение, связанные с использованием неутвержденного препарата. Medicare и Medicaid не оплачивают лекарства, которые не одобрены FDA или не включены в компендиум, а оплачивают только лечение, связанное с введением исследуемых агентов в контексте квалификационного исследования. Таким образом, ни Medicare, ни Medicaid в соответствии с настоящим Законом не предназначены для покрытия расходов на неутвержденный препарат, связанных с ним услуг по приему этого препарата или лечения каких-либо медицинских последствий применения этого препарата. Часто коммерческое страхование следует примеру Medicare. Таким образом, оплата медицинской помощи в соответствии с этим Законом, даже если фармацевтическая компания будет бесплатно предоставлять исследуемый препарат, может представлять собой значительное препятствие. Конкретное заявление здесь о том, что этот Закон «не устанавливает положительного права для какой-либо стороны или лица», подчеркивает, что законодательство не наделяет пациента явным «правом на судебное разбирательство».
Следует отметить, что в соответствии с настоящим Законом не существует конкретных указаний относительно того, кто покрывает стоимость препарата или расходы на лечение, связанные с использованием неутвержденного препарата. Medicare и Medicaid не оплачивают лекарства, которые не одобрены FDA или не включены в компендиум, а оплачивают только лечение, связанное с введением исследуемых агентов в контексте квалификационного исследования. Таким образом, ни Medicare, ни Medicaid в соответствии с настоящим Законом не предназначены для покрытия расходов на неутвержденный препарат, связанных с ним услуг по приему этого препарата или лечения каких-либо медицинских последствий применения этого препарата. Часто коммерческое страхование следует примеру Medicare. Таким образом, оплата медицинской помощи в соответствии с этим Законом, даже если фармацевтическая компания будет бесплатно предоставлять исследуемый препарат, может представлять собой значительное препятствие. Конкретное заявление здесь о том, что этот Закон «не устанавливает положительного права для какой-либо стороны или лица», подчеркивает, что законодательство не наделяет пациента явным «правом на судебное разбирательство».
Кроме того, в следующем пункте этого раздела говорится, что «сенат считает, что… [данный закон] не устанавливает никаких новых мандатов, директив или дополнительных правил», подтверждая, что этот закон не требует каких-либо отдельных или юридическое лицо для предоставления исследуемого препарата в соответствии с настоящим Законом.
Наконец, пункт номер пять предлагает предостережение тем лицам, которые в состоянии эмоционального стресса, связанного с преодолением неизлечимой болезни, могут утратить представление о том, что могут и чего не могут делать агенты расследования: «Сенат считает, что … [этот закон] не будет и не может создать лекарство или эффективную терапию там, где их не существует».
Полезно рассмотреть, что остается открытым для интерпретации в RTT, что не включено и чем RTT отличается от программ расширенного доступа или благотворительного использования FDA. В RTT нет рекомендаций по мониторингу данных или последующему наблюдению за каждым отдельным пациентом. Хотя производитель должен предоставить отчет в FDA, как указано выше, сбор и передача данных от лечащего врача производителю остается на усмотрение этих сторон. Лечащий врач не предъявляет требований к отчетности в FDA. Единственная роль FDA в RTT ограничивается получением годового отчета от производителя и размещением сводного отчета на их веб-сайте.
Хотя производитель должен предоставить отчет в FDA, как указано выше, сбор и передача данных от лечащего врача производителю остается на усмотрение этих сторон. Лечащий врач не предъявляет требований к отчетности в FDA. Единственная роль FDA в RTT ограничивается получением годового отчета от производителя и размещением сводного отчета на их веб-сайте.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) не участвует в рассмотрении или утверждении пациента, получающего исследуемый препарат в рамках RTT. Если подходящий пациент ищет подходящее лекарство, определение приемлемости осуществляется исключительно между пациентом, лечащим врачом и производителем. Это значительно отличается от программы расширенного доступа FDA, в которой лечащие врачи подают форму FDA 3926 для индивидуального расширенного доступа к исследуемым лекарственным средствам и биологическим препаратам, или формы FDA 1571 и 1572 требуются для других IND лечения промежуточного доступа и отраслевых IND. Кроме того, производители, которые соглашаются предоставлять лекарства через расширенный доступ, должны разрешить FDA ссылаться на их отраслевой IND для этого препарата, предоставив FDA письмо-разрешение, или они должны поддержать запрос, предоставив лечащему врачу информацию о медицинском продукте. . При расширенном доступе или сострадательном использовании FDA должно просмотреть все формы запросов, а затем определить, можно ли продолжить лечение. 4 В RTT нет такого нормативного требования.
Кроме того, производители, которые соглашаются предоставлять лекарства через расширенный доступ, должны разрешить FDA ссылаться на их отраслевой IND для этого препарата, предоставив FDA письмо-разрешение, или они должны поддержать запрос, предоставив лечащему врачу информацию о медицинском продукте. . При расширенном доступе или сострадательном использовании FDA должно просмотреть все формы запросов, а затем определить, можно ли продолжить лечение. 4 В RTT нет такого нормативного требования.
В отличие от роли Институционального контрольного совета (IRB) в отношении исследуемых препаратов с расширенным доступом, в котором IRB уполномочен рассматривать протоколы расширенного доступа и формы согласия, IRB не играет какой-либо конкретной роли в рамках RTT. Никакие формы не должны быть представлены или одобрены ЭСО в рамках RTT, и ЭСО не имеет никаких обязательств в отношении введения лекарственного средства в соответствии с механизмом RTT. Хотя RTT требует, чтобы врач получил письменное информированное согласие, он не определяет требования к такому согласию и не требует, чтобы ЭСО рассматривал или утверждал форму согласия.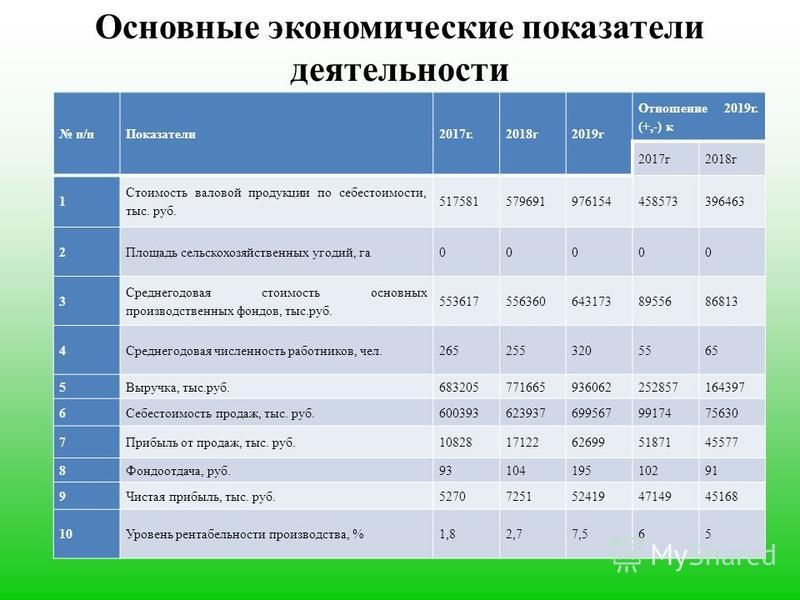
RTT прямо не указывает, кто определяет, соблюдаются ли требования настоящего Закона. Как написано в настоящее время, похоже, что эти обязанности принадлежат производителю, учитывая, что компания по-прежнему должна предоставлять общую информацию FDA относительно количества доз, количества пациентов и побочных эффектов, связанных с конкретным лекарством, предоставляемым в соответствии с этим Законом. . Процесс определения пригодности пациента и лекарственного препарата, особенности формы информированного согласия и предоставление данных должны согласовываться как производителем, так и лечащим врачом в каждом конкретном случае, и, как гласит закон, ни лечащий врач, ни производитель несет ответственность за введение или предоставление препарата. Также следует отметить, что в RTT не упоминается публикация результатов, поэтому нет указаний относительно того, могут ли быть опубликованы результаты пациентов, получавших RTT, или нет.
RTT создает для пациентов возможность получения исследуемого препарата вне клинических испытаний, если и только если пациент соответствует критериям, препарат соответствует критериям, и пациент, производитель препарата и лечащий врач согласны с тем, что они желают идти по этому пути. Это законодательство не дает пациенту права принуждать врача или фармацевтическую компанию предоставлять лекарство в соответствии с этим Законом. Чтобы пациент соответствовал критериям, у него должно быть опасное для жизни заболевание или состояние, он должен исчерпать стандартные варианты лечения и не должен иметь доступа к лекарству, проходящему клиническое испытание. Чтобы лекарство было одобрено, оно должно быть , а не , должны быть одобрены FDA для любых показаний, должны быть завершены испытания фазы I и должны иметь продолжающиеся базовые испытания. Количество случаев, когда все критерии приемлемости для пациента и препарата соблюдены, и все стороны соглашаются продолжить, будет ограничено, однако в таких сценариях RTT обеспечивает путь, который намного проще и требует гораздо меньше консультаций, документации , и отчетность, чем Программа расширенного доступа FDA, тем самым облегчая доступ к подходящим исследуемым агентам для подходящих пациентов.
Это законодательство не дает пациенту права принуждать врача или фармацевтическую компанию предоставлять лекарство в соответствии с этим Законом. Чтобы пациент соответствовал критериям, у него должно быть опасное для жизни заболевание или состояние, он должен исчерпать стандартные варианты лечения и не должен иметь доступа к лекарству, проходящему клиническое испытание. Чтобы лекарство было одобрено, оно должно быть , а не , должны быть одобрены FDA для любых показаний, должны быть завершены испытания фазы I и должны иметь продолжающиеся базовые испытания. Количество случаев, когда все критерии приемлемости для пациента и препарата соблюдены, и все стороны соглашаются продолжить, будет ограничено, однако в таких сценариях RTT обеспечивает путь, который намного проще и требует гораздо меньше консультаций, документации , и отчетность, чем Программа расширенного доступа FDA, тем самым облегчая доступ к подходящим исследуемым агентам для подходящих пациентов.
Клинико-трансляционная значимость
Больные раком, которые хотят получить исследуемые препараты, слишком часто не могут получить к ним доступ через участие в клиническом испытании. Причины могут включать в себя отсутствие доступа к испытанию, отсутствие свободного места для участия в испытании или несоблюдение одного или нескольких квалификационных требований. Закон о праве на судебное разбирательство — это один из механизмов, который могут использовать пациенты и врачи, желающие получить доступ к исследуемому лекарственному средству вне испытаний. Тем не менее, этот закон не очень хорошо известен и не очень хорошо понимается в онкологическом сообществе. Мы рассматриваем это федеральное законодательство и обсуждаем, для каких препаратов и для каких пациентов оно может быть применимо, а также где оно применимо, а где нет, может облегчить доступ пациентов к многообещающим экспериментальным методам лечения вне исследования. Мы также сравниваем и противопоставляем его имеющимся программам расширенного доступа и благотворительного использования в рамках FDA и обсуждаем соответствующие различия.
Причины могут включать в себя отсутствие доступа к испытанию, отсутствие свободного места для участия в испытании или несоблюдение одного или нескольких квалификационных требований. Закон о праве на судебное разбирательство — это один из механизмов, который могут использовать пациенты и врачи, желающие получить доступ к исследуемому лекарственному средству вне испытаний. Тем не менее, этот закон не очень хорошо известен и не очень хорошо понимается в онкологическом сообществе. Мы рассматриваем это федеральное законодательство и обсуждаем, для каких препаратов и для каких пациентов оно может быть применимо, а также где оно применимо, а где нет, может облегчить доступ пациентов к многообещающим экспериментальным методам лечения вне исследования. Мы также сравниваем и противопоставляем его имеющимся программам расширенного доступа и благотворительного использования в рамках FDA и обсуждаем соответствующие различия.
R. Agarwal и L. Saltz частично при поддержке: P30-17 CA008748.
Р. Агарвал частично поддерживается Фондом борьбы с раком ASCO. Эта работа финансировалась премией Анны Браглиа для молодых исследователей в области поддерживающей терапии рака, поддерживаемой HELSINN. Любые мнения, выводы и заключения, выраженные в этом материале, принадлежат автору (авторам) и не обязательно отражают точку зрения Американского общества клинической онкологии® или фонда Conquer Cancer Foundation.
Агарвал частично поддерживается Фондом борьбы с раком ASCO. Эта работа финансировалась премией Анны Браглиа для молодых исследователей в области поддерживающей терапии рака, поддерживаемой HELSINN. Любые мнения, выводы и заключения, выраженные в этом материале, принадлежат автору (авторам) и не обязательно отражают точку зрения Американского общества клинической онкологии® или фонда Conquer Cancer Foundation.
| RTT | Закон о праве на судебное разбирательство |
| FDA | Управление по санитарному надзору за качеством пищевых продуктов и медикаментов |
Авторы заявляют об отсутствии потенциального конфликта интересов.
1. [Проверено 10.02.19]; https://www.congress.gov/115/bills/s204/BILLS-115s204enr.pdf.
2. [по состоянию на 10 февраля 2019 г.]; https://uscode.house.gov/browse/prelim@title21/chapter9/subchapter5/partE&edition=prelim.
3. [проверено 10-2-19]; https://ecfr. io/Title-21/cfr312_main.
io/Title-21/cfr312_main.
4. Jarow JP, et al. (2017). «Десятилетний опыт работы Центра оценки и исследования лекарственных средств, часть 2: роль FDA в обеспечении безопасности пациентов». The Innov Regul Sci
51(2): 246–249. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Пристальный взгляд на новый федеральный закон «О праве на судебное разбирательство»
30 мая 2018 г. четырехлетнее движение «Право на судебное разбирательство» (RTT) завершилось принятием нового федерального закона, заявленной целью которого является оказание помощи неизлечимо больным пациентам. с новым путем доступа к исследуемым препаратам ранней фазы, которые в настоящее время находятся на стадии клинической разработки, не одобрены или не доступны для всех нуждающихся пациентов.
Законы RTT направлены на устранение надзора и правил Федерального управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) в процессе предварительного одобрения, в котором пациенты стремятся получить доступ к экспериментальным лекарствам, находящимся в разработке, вне клинических испытаний, в которых лекарства проходят испытания, 1 , несмотря на то, что неизлечимо больные пациенты десятилетиями имели право запрашивать такой доступ в соответствии с правилами FDA в рамках Программы расширенного доступа (EAP).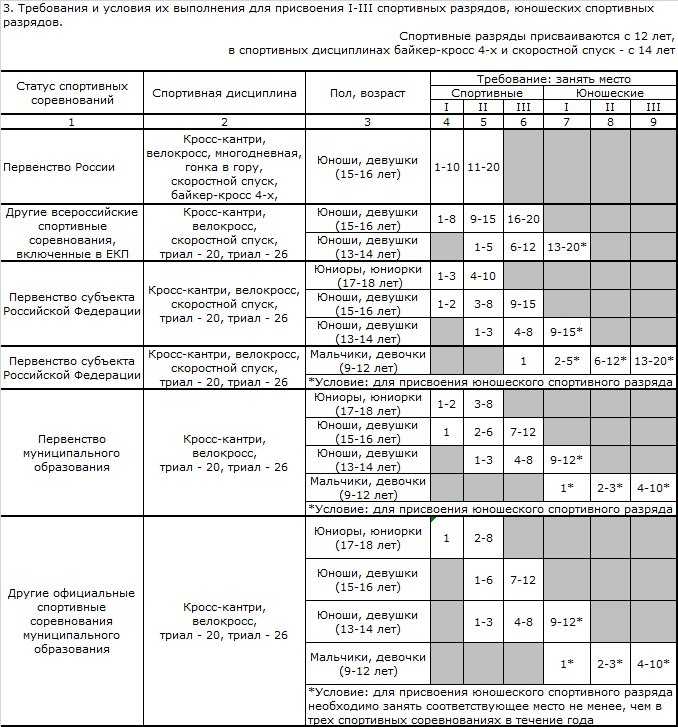 Действительно, тысячи пациентов успешно получили доступ в рамках механизма EAP; FDA одобряет почти 99% всех запросов на благотворительное использование, 2 обычно примерно через три дня. Также были предприняты дополнительные инициативы FDA по упорядочению и упрощению процесса EAP как для спонсоров, так и для запрашивающих врачей, а также для улучшения понимания общественностью федеральных механизмов, позволяющих пациентам запрашивать предварительный доступ. (Дополнительные доступные инструменты включают расширенный доступ FDA: страница «Информация для промышленности» и навигатор расширенного доступа.)
Действительно, тысячи пациентов успешно получили доступ в рамках механизма EAP; FDA одобряет почти 99% всех запросов на благотворительное использование, 2 обычно примерно через три дня. Также были предприняты дополнительные инициативы FDA по упорядочению и упрощению процесса EAP как для спонсоров, так и для запрашивающих врачей, а также для улучшения понимания общественностью федеральных механизмов, позволяющих пациентам запрашивать предварительный доступ. (Дополнительные доступные инструменты включают расширенный доступ FDA: страница «Информация для промышленности» и навигатор расширенного доступа.)
Несмотря на свое название, законы о РТТ не предоставляют фактического «права» на получение доступа к лекарству, находящемуся в разработке, по запросу; они дают только право на запрос доступа — запрос, который уже разрешен текущими правилами FDA. Нет включенного мандата на предоставление экспериментального продукта по запросу пациента, оставляя важнейшие аспекты доступа на усмотрение спонсоров, производителей и учреждений. Вместо такого мандата законы содержат множество положений, направленных на то, чтобы сделать практическую среду, связанную с предоставлением доступа к предварительному утверждению, более благоприятной, в надежде, что это побудит спонсоров и других лиц удовлетворять запросы. Эти положения, однако, не только лишают пациентов важнейших гарантий, но и могут привести к нанесению пациенту вреда, обращение за помощью в отношении которого запрещено законом.
Вместо такого мандата законы содержат множество положений, направленных на то, чтобы сделать практическую среду, связанную с предоставлением доступа к предварительному утверждению, более благоприятной, в надежде, что это побудит спонсоров и других лиц удовлетворять запросы. Эти положения, однако, не только лишают пациентов важнейших гарантий, но и могут привести к нанесению пациенту вреда, обращение за помощью в отношении которого запрещено законом.
Несмотря на эти факты и статистику, сторонники подхода RTT ухватились за очень деликатные вопросы, связанные с неизлечимыми заболеваниями, включая право умирающих пациентов выбирать, как им провести оставшиеся дни, и разочарование длительными сроками разработки и утверждения лекарств, чтобы успешно нацелены на FDA как на главное препятствие для доступа пациентов к потенциально спасающим жизнь методам лечения. Успех этого подхода привел не только к принятию нового федерального закона, но и к тому, что 41 штат США принял законы о RTT, начиная с мая 2014 года. подтвержденный случай получения пациентом доступа к исследовательскому вмешательству исключительно в соответствии с законом штата о RTT.
подтвержденный случай получения пациентом доступа к исследовательскому вмешательству исключительно в соответствии с законом штата о RTT.
Это может измениться с принятием федерального закона о RTT, хотя, скорее всего, не в том положительном ключе, как следует из риторики вокруг закона.
Находясь на высоком уровне, федеральный закон о РТ использует тот же теоретический подход к «решению» «проблемы» доступа до утверждения, что и законы штата о РТТ, например, устранение предполагаемых барьеров для доступа, создаваемых FDA и его применимыми правилами. и правила для доступа к предварительному разрешению — язык федерального закона не просто отражает амбициозные, но неэффективные законы штата о RTT, которые, как многие утверждали, не более чем дают пациентам ложную надежду. 3 Вместо этого федеральный закон о RTT фактически эффективно манипулирует юрисдикцией FDA, создавая альтернативный дополнительный путь для частных лиц, чтобы запросить ранний доступ. В дополнение к удалению FDA и ключевых правил надзора из процесса предварительного утверждения, это устраняет необходимость надзора IRB, изолирует спонсоров, производителей и других от ответственности, возникающей в результате вреда, причиненного доступом, позволяет страховщикам снизить любые расходы, связанные с введение экспериментальной терапии и, возможно, изменяет основные требования к отчетности по клиническим исходам по сравнению с предварительным доступом.
В дополнение к удалению FDA и ключевых правил надзора из процесса предварительного утверждения, это устраняет необходимость надзора IRB, изолирует спонсоров, производителей и других от ответственности, возникающей в результате вреда, причиненного доступом, позволяет страховщикам снизить любые расходы, связанные с введение экспериментальной терапии и, возможно, изменяет основные требования к отчетности по клиническим исходам по сравнению с предварительным доступом.
Все это заставило критиков задаться вопросом, что именно получили пациенты в соответствии с этим новым федеральным законом о RTT. В то же время спонсоры, исследовательские институты, врачи и государственные органы пытаются согласовать положения нового закона с существующими федеральными нормами и правилами.
Чем доступ RTT отличается от доступа в соответствии с действующим федеральным законодательством?
Новый федеральный закон RTT вносит поправки в Федеральный закон о пищевых продуктах, лекарствах и косметических средствах, чтобы предоставить лицам дополнительный новый способ запросить предварительное разрешение на доступ к исследуемым препаратам при условии, что у них диагностировано опасное для жизни состояние или заболевание, исчерпали уже утвержденные методы лечения и не могут участвовать в клинических испытаниях соответствующего исследуемого препарата. Эти требования должны быть подтверждены сертифицирующим врачом, который не может получать компенсацию напрямую от производителя препарата за эту сертификацию.
Эти требования должны быть подтверждены сертифицирующим врачом, который не может получать компенсацию напрямую от производителя препарата за эту сертификацию.
В качестве двух путей, предназначенных для предоставления неизлечимо больным пациентам доступа к экспериментальным препаратам, которые находятся в стадии изучения, но еще не одобрены FDA, существует некоторое сходство между расширенным доступом и альтернативным путем, созданным в соответствии с федеральным законом RTT. Но в новом законе также есть ключевые отличия и неясности, которые вызывают серьезные опасения с точки зрения защиты и безопасности пациентов:
- Федеральный закон о РТТ прямо отменяет подавляющее большинство частей 50, 56 и 312 главы 21 Кодекса. Федеральных правил (CFR), которые содержат правила и положения для надзора IRB за тестированием на наркотики, включая юрисдикцию для расширенных запросов на доступ и требования информированного согласия. Действительно, в федеральном законе о РТ (в отличие от некоторых законов штатов) не упоминается ни ЭСО, ни этическая экспертиза, ни даже проверка какой-либо независимой третьей стороной (например, местным правительственным или институциональным органом).
 Кроме того, в то время как закон о RTT гласит, что пациенты или их законные представители должны предоставить «информированное согласие» лечащему врачу на использование исследуемого препарата, закон о RTT прямо освобождает доступ к RTT из всех положений об информированном согласии, содержащихся в федеральных правилах на 21 CFR 50, которые применимы к запросам в рамках EAP. Закон не заменяет эти ключевые детали какими-либо альтернативными требованиями, в том числе в отношении достоверности передаваемой информации, добровольности предоставленного согласия или проверки процедур информированного согласия со стороны ЭСО или другой третьей стороны.
Кроме того, в то время как закон о RTT гласит, что пациенты или их законные представители должны предоставить «информированное согласие» лечащему врачу на использование исследуемого препарата, закон о RTT прямо освобождает доступ к RTT из всех положений об информированном согласии, содержащихся в федеральных правилах на 21 CFR 50, которые применимы к запросам в рамках EAP. Закон не заменяет эти ключевые детали какими-либо альтернативными требованиями, в том числе в отношении достоверности передаваемой информации, добровольности предоставленного согласия или проверки процедур информированного согласия со стороны ЭСО или другой третьей стороны. - В соответствии с законом RTT исследуемый препарат, на который может быть подана заявка, также должен соответствовать определенным требованиям, неразрывно связанным с действующим федеральным законодательством, включая то, что он является предметом заявки, поданной в FDA (в соответствии со статьей 505(b) или 351(a) Закон об общественном здравоохранении). Исследуемые препараты имеют право на участие только в том случае, если они уже прошли клиническое исследование фазы I; не одобрены FDA для любых показаний; являются частью либо находящейся на рассмотрении заявки FDA на одобрение лекарственного средства, либо текущего испытания, данные которого будут представлены в FDA как часть заявки; и не были поставлены на клиническую паузу во время разработки. 4 Эти требования могут обеспечить базовый уровень безопасности и, по крайней мере, некоторую уверенность в отношении рассматриваемых препаратов. Однако они также исключают доступ к некоторым ранним соединениям, которые могут быть разрешены FDA для отдельных пациентов в соответствии с федеральным регламентом.
Возможно, это обеспечивает некоторый противовес отмене федеральных мер защиты, включенных в существующий процесс расширенного доступа. Однако расплывчатые формулировки и неопределенные термины в законе о RTT создают ряд потенциальных лазеек. К ним относятся отсутствие какого-либо требования о том, чтобы организация, предоставляющая доступ к препарату по альтернативному пути, была той же самой организацией, которая является спонсором поданной заявки на получение нового исследуемого препарата (IND), и любой независимой независимой проверки (вместо FDA или даже IRB), чтобы обеспечить уровень целостности или значимости «завершенного» исследования фазы 1.
- В отличие от пути EAP, федеральный закон о RTT указывает, что клинические исходы, связанные с использованием подходящего исследуемого препарата, как правило, не могут быть использованы для неблагоприятного воздействия на рассмотрение или одобрение этого препарата, хотя все известные серьезные нежелательные явления (СНЯ) должны быть обобщены. в годовом отчете и подается вместе с новой заявкой на препарат. Кроме того, в законе о RTT четко указано, что спонсор, производитель или другой «распространитель» исследуемого препарата не несет ответственности за какое-либо действие или бездействие в отношении приемлемого исследуемого препарата. Это последнее положение противоречит федеральным правилам, касающимся информированного согласия на расширенный доступ, которые прямо запрещают включение оправдательных формулировок.
 Кроме того, в то время как закон о RTT гласит, что пациенты или их законные представители должны предоставить «информированное согласие» лечащему врачу на использование исследуемого препарата, закон о RTT прямо освобождает доступ к RTT из всех положений об информированном согласии, содержащихся в федеральных правилах на 21 CFR 50, которые применимы к запросам в рамках EAP. Закон не заменяет эти ключевые детали какими-либо альтернативными требованиями, в том числе в отношении достоверности передаваемой информации, добровольности предоставленного согласия или проверки процедур информированного согласия со стороны ЭСО или другой третьей стороны.
Кроме того, в то время как закон о RTT гласит, что пациенты или их законные представители должны предоставить «информированное согласие» лечащему врачу на использование исследуемого препарата, закон о RTT прямо освобождает доступ к RTT из всех положений об информированном согласии, содержащихся в федеральных правилах на 21 CFR 50, которые применимы к запросам в рамках EAP. Закон не заменяет эти ключевые детали какими-либо альтернативными требованиями, в том числе в отношении достоверности передаваемой информации, добровольности предоставленного согласия или проверки процедур информированного согласия со стороны ЭСО или другой третьей стороны.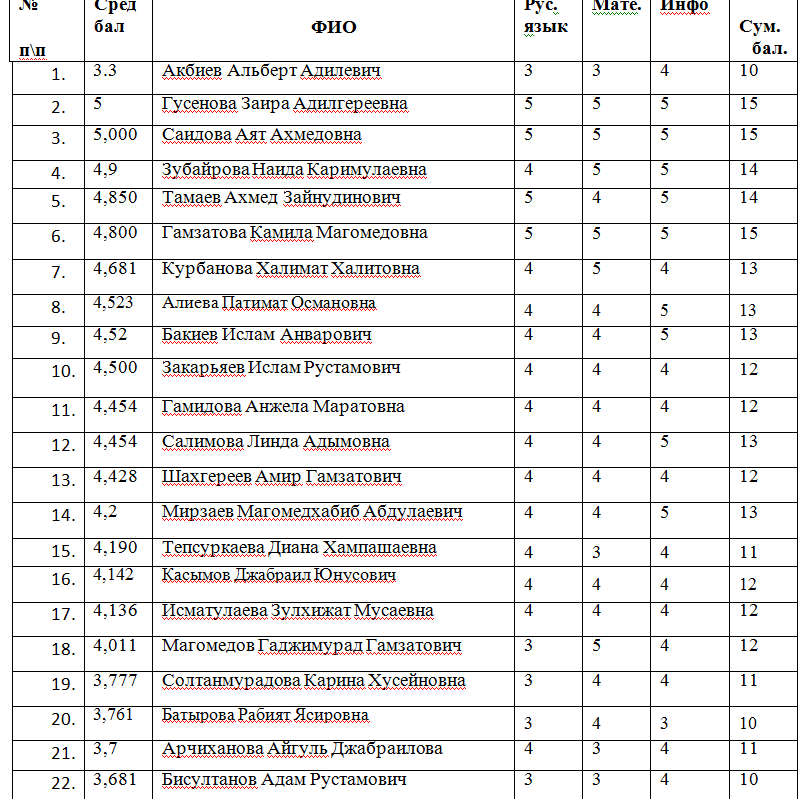 Исследуемые препараты имеют право на участие только в том случае, если они уже прошли клиническое исследование фазы I; не одобрены FDA для любых показаний; являются частью либо находящейся на рассмотрении заявки FDA на одобрение лекарственного средства, либо текущего испытания, данные которого будут представлены в FDA как часть заявки; и не были поставлены на клиническую паузу во время разработки. 4 Эти требования могут обеспечить базовый уровень безопасности и, по крайней мере, некоторую уверенность в отношении рассматриваемых препаратов. Однако они также исключают доступ к некоторым ранним соединениям, которые могут быть разрешены FDA для отдельных пациентов в соответствии с федеральным регламентом.
Исследуемые препараты имеют право на участие только в том случае, если они уже прошли клиническое исследование фазы I; не одобрены FDA для любых показаний; являются частью либо находящейся на рассмотрении заявки FDA на одобрение лекарственного средства, либо текущего испытания, данные которого будут представлены в FDA как часть заявки; и не были поставлены на клиническую паузу во время разработки. 4 Эти требования могут обеспечить базовый уровень безопасности и, по крайней мере, некоторую уверенность в отношении рассматриваемых препаратов. Однако они также исключают доступ к некоторым ранним соединениям, которые могут быть разрешены FDA для отдельных пациентов в соответствии с федеральным регламентом.
Последствия и последствия
Отсутствие в федеральном законе о РТТ мандата на предоставление раннего доступа к исследуемому препарату по запросу пациента сохраняет свободу действий производителей лекарств и других спонсоров в предоставлении раннего доступа, аналогичную той, которой обладают спонсоры. в соответствии с действующим федеральным законодательством. Если спонсор или производитель желает предоставить ранний доступ физическому лицу, при условии соблюдения всех других требований, у организаций есть возможность использовать альтернативный путь, предусмотренный федеральным законом RTT 9.0013 или в соответствии с действующим федеральным законодательством о расширенном доступе.
в соответствии с действующим федеральным законодательством. Если спонсор или производитель желает предоставить ранний доступ физическому лицу, при условии соблюдения всех других требований, у организаций есть возможность использовать альтернативный путь, предусмотренный федеральным законом RTT 9.0013 или в соответствии с действующим федеральным законодательством о расширенном доступе.
Действительно, за то короткое время, что новый федеральный закон был в книгах, несколько крупных фармацевтических компаний публично заявили о своем намерении продолжать придерживаться существующих федеральных правил для индивидуального расширенного доступа и с участием FDA, а не следовать RTT. путь. Однако, независимо от размера компании и других атрибутов, ясно, что спонсоры и производители уделяют много времени и внимания формированию политики и стратегии в этой области. 5
Несмотря на то, что потенциальное влияние RTT на организации, проводящие клинические испытания, мало обсуждалось общественностью, академические медицинские центры, центры клинических испытаний и IRB, возможно, должны противостоять и урегулировать противоречивые обязанности между традиционными федеральными требованиями к клиническим испытаниям и расширенный доступ и новая федеральная структура RTT (например, конфликт между защитой RTT спонсоров и производителей от ответственности и стандартными федеральными требованиями в отношении информированного согласия, запрещающими оправдательные формулировки).
Хотя это прямо не указано в законе, учреждения, похоже, имеют право решать, будут ли они следовать новым правилам (или, возможно, отсутствию правил) в соответствии с RTT, и если да, то каким образом. Как и в случае со спонсорами, ничто в законе о RTT не запрещает учреждениям создавать единый набор последовательных правил для запросов на доступ к предварительному разрешению, которые тесно связаны с текущими правилами и положениями FDA, например, требуя рассмотрения IRB и элементов информированного согласия, даже для запросов в рамках RTT. Учреждения могут сделать это по ряду причин, в том числе для обеспечения согласованности надзора за проведением клинических испытаний, защиты пациентов, избежания рисков и других этических и практических соображений.
Будем рады услышать от вас!
Обсуждалось ли в вашем учреждении новый закон о RTT и как вы будете обрабатывать запросы на RTT? Как будет участвовать ваш IRB? Если бы вы создали какие-либо ресурсы, хотели бы вы поделиться ими с сообществом? Комментарий ниже.
Beth E. Roxland, JD, MBioethics — адвокат и специалист по биоэтике с уникальным опытом работы в промышленности, юриспруденции, правительстве и научных кругах. Она является старшим советником по праву, политике в области здравоохранения и этике в компании Roxland Consultants, Ltd., консультирующей юридические фирмы, медико-биологические учреждения, а также профессиональные ассоциации и ассоциации пациентов. Она часто читает лекции и публикуется по различным передовым нормативным и биоэтическим темам, а также входит в несколько научных и академических наблюдательных советов в ведущих учреждениях. Г-жа Роксленд ранее была руководителем отдела биоэтики и стратегии Johnson & Johnson в офисе главного врача. Она также была исполнительным директором Целевой группы штата Нью-Йорк по вопросам жизни и права и одновременно работала специальным советником комиссара здравоохранения по этике исследований стволовых клеток. До прихода в правительство г-жа Роксланд была старшим юристом по судебным разбирательствам в Simpson Thacher & Bartlett LLP и клерком федерального судебного права в Южном округе Нью-Йорка. Она окончила Колумбийский университет со степенью бакалавра биологии и с отличием получила совместную степень доктора права и магистра биоэтики в Пенсильванском университете, где работала старшим редактором журнала Law Review Пенсильванского университета.
Она окончила Колумбийский университет со степенью бакалавра биологии и с отличием получила совместную степень доктора права и магистра биоэтики в Пенсильванском университете, где работала старшим редактором журнала Law Review Пенсильванского университета.
Элиза А. Херли, доктор философии , является исполнительным директором PRIM&R. Найдите ее биографию на сайте PRIM&R.
7 марта 2018 г. PRIM&R провела вебинар под названием Доступ к лекарственным препаратам на этапе предварительного утверждения: ориентируясь в меняющейся нормативно-правовой, этической и правовой среде HR руководство, недавнее законодательство и этические вопросы, связанные с расширенным доступом и правом на попытку. Бет Роксленд была соведущей с Ричард Кляйн на этом вебинаре. Запись вебинара доступна для покупки физическими лицами в интернет-магазине . Если вы хотите приобрести вебинар для группового просмотра, загрузите форму заказа (PDF) и отправьте по адресу Registration@primr. org . С г-жой Роксланд также можно связаться по вопросам по адресу [email protected].
org . С г-жой Роксланд также можно связаться по вопросам по адресу [email protected].
[1] Джонатан П. Джароу, и др. Обзор программы расширенного доступа FDA к исследуемым препаратам , ## Терапевтические инновации и нормативная наука ## (2017). См. также Emily Jung, Patricia J. Zettler, & Aaron S. Kesselheim, Преобладание общедоступных политик расширенного доступа , ## Clin. Pharmacology & Therapeutics ## (13 декабря 2018 г.)
[2] Amy E. McKee, et. номер . Как часто лекарства становятся доступными в соответствии с утвержденным процессом расширенного доступа Управления по санитарному надзору за качеством пищевых продуктов и медикаментов? 57 J. Clin. Фармакология (2017)
[3] Бет Роксланд, Пациенты заслуживают лучшего, чем «Право на [надежду] попробовать », The Hill (август 2016 г.), , доступно по телефону http://thehill.com/blogs/congress-blog/ здравоохранение/2